Abstract
The Hippo pathway plays an important role in the growth, development, and regeneration of cells and organs. Transcriptional enhanced associate domain (TEAD), a transcription activator of the Hippo pathway, forms the complex with a transcriptional coactivator yes-associated protein (YAP) or a transcriptional coactivator PDZ-binding motif (TAZ). Their excessive activations are involved in carcinogenesis such as malignant pleural mesothelioma (MPM), and thus inhibition of the TEAD complex is expected to have potent anticancer activity against MPM. On the other hand, YAP or TAZ conditional knockout mice have been reported to show abnormal findings in various tissues, including the kidney, liver, and lung. In the present study, we evaluated the systemic toxicity of K-975, a novel TEAD inhibitor, in rats. When K-975 was administered orally to rats for 1 week, proteinuria suggestive of nephrotoxicity was observed. Electron microscopy revealed that K-975 at 300 mg/kg induced glomerular podocyte foot process effacement. After a 2-week recovery period, proteinuria with foot process effacement was recovered completely. Urinalysis and urinary biomarker evaluation suggested that the urinary albumin index (urinary albumin/urinary creatinine) was the most sensitive marker for detecting K-975-induced nephrotoxicity. After 3 cycles of 1-week administration followed by 2-week recovery periods, nephrotoxicity was reversible; however, incomplete reversibility was observed in rats with severe proteinuria. In conclusion, this study revealed that in rats, oral K-975 treatment induced severe proteinuria by podocyte foot process effacement, which was reversible and monitorable by the urinary albumin index, suggesting important information for developing K-975 as an anticancer drug.
INTRODUCTION
The Hippo pathway, originally identified in Drosophila and highly conserved in mammals (Justice et al., 1995), plays an important role in cell and organ growth, biological development, regeneration, and homeostasis (Dupont et al., 2011; Rausch and Hansen, 2020; Park and Hansen, 2021). The mammalian Hippo pathway is composed of molecules, including neurofibromin 2 (NF2), mammalian sterile 20-like kinase 1/2 (MST1/2) and large tumor suppressor kinase 1/2 (LATS1/2). Upon activation, these molecules promote the phosphorylation of downstream effectors of the Hippo pathway, yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), leading to their ubiquitination-dependent degradation (Piccolo et al., 2014). On the other hand, dephosphorylated YAP and TAZ, which act as transcriptional coactivators, translocate into the nucleus to form a complex with transcriptional enhanced associate domain (TEAD), which is a DNA binding transcription factor (Piccolo et al., 2014). The complex then activates the transcription of genes, such as ankyrin repeat domain-containing protein (ANKRD), cysteine-rich 61 (CYR61), and connective tissue growth factor (CTGF) for cell proliferation and embryonic and organ development (Haskins et al., 2014; Wang et al., 2016).
The Hippo pathway and its downstream molecules are very important due to their biologically fundamental roles; however, excessive activation of downstream molecules, YAP, TAZ, and TEAD, is known to be involved in carcinogenesis. The knockout of Nf2, Lats1/2, or Mst1/2 in mice is known to cause osteosarcoma and hepatocellular carcinoma (McClatchey et al., 1998), soft tissue sarcomas and ovarian tumors (St John et al., 1999), or cholangiocarcinoma and colonic adenoma (Zhou et al., 2009; Lu et al., 2010; Song et al., 2010), respectively. YAP, TAZ, and TEAD are also known as oncogenes and are overexpressed in various human cancers, such as malignant mesothelioma (Fujii et al., 2012), ovarian cancer (Zhang et al., 2011), cholangiocarcinoma (Pei et al., 2015), hepatocellular and squamous cell carcinomas, gastrointestinal dysplasia (Wong et al., 2016; Zhou et al., 2016), and pancreatic duct metaplasia (Camargo et al., 2007). These malignant tumors and precancerous changes are attributed to overactivation of TEAD-dependent gene transcription, which enhances cell proliferation, metastasis, epithelial to mesenchymal transition, and cancer stem cell maintenance in tumor cells (Moroishi et al., 2015). Therefore, TEAD plays a crucial role in cancer progression, and TEAD inhibitors are expected to be developed as anticancer agents.
K-975 is a novel TEAD inhibitor (Kaneda et al., 2020). This compound has been reported to exhibit antitumor activity against malignant pleural mesothelioma (MPM) in both in vitro cultured cells and mouse xenograft models in vivo. Additionally, treatment with a combination of K-975 and pemetrexed, cisplatin, or palbociclib is associated with higher survival rates and antitumor activities in comparison to monotherapy with K-975. Therefore, K-975 is expected to be an anticancer drug for MPM not only as a monotherapy, but also as a combination therapy with standard treatment. The reason why K-975 exhibits potent anticancer activity is that it inhibits the protein-protein interaction between YAP/TAZ and TEAD by covalently binding to an internal cysteine residue located in the palmitate-binding pocket of TEAD, which is essential for palmitoylation of TEAD to improve the binding affinity to YAP/TAZ and form the stable YAP/TAZ-TEAD complex (Chan et al., 2016; Noland et al., 2016). In addition to K-975, several TEAD inhibitors have been reported to similarly target this palmitate-binding pocket (Cunningham and Hansen, 2022). Therefore, TEAD inhibitors, including K-975, may be used as novel anticancer drugs in the near future.
However, some safety concerns have been reported regarding TEAD inhibition. Mice with the germline deletion of murine Yap, Tead1, or Tead4 exhibit embryonic lethality (Schlegelmilch et al., 2011; Kakiuchi-Kiyota et al., 2019). Mice with conditional KO (cKO) of Yap or Taz show increased liver size and liver injury (Lu et al., 2018), thinned ventricular walls, and dilated cardiomyopathy (Xin et al., 2013), and abnormal phenotypes associated with lung development (Makita et al., 2008). In particular, the effects of the deletion of YAP or TAZ in the kidney have been well studied (Wong et al., 2016). Yap cKO mice and Taz KO mice showed defects in kidney organogenesis (Hossain et al., 2007; Makita et al., 2008; Reginensi et al., 2013), and podocyte-specific Yap deficiency resulted in focal segmental glomerulosclerosis, proteinuria, and renal failure (Schwartzman et al., 2016). These reports suggest that TEAD inhibition-mediated toxicity, particularly nephrotoxicity, could be a toxicological concern in the development of TEAD inhibitors as therapeutic drugs. However, the roles that YAP/TAZ and TEAD play in adult tissues remain unclear (Kaneda et al., 2020).
Podocytes, a potential toxic target for TEAD inhibitors, are terminally differentiated cells in the glomerulus and retain characteristic morphology, foot processes. The podocyte foot processes interdigitate with neighboring cells, forming a network that encapsulates the glomerular capillaries. These foot processes play an important role in the filtration of blood into urine, especially in preventing the leakage of macromolecules into the urine, such as albumin (Pavenstädt et al., 2003; Reiser and Altintas, 2016). After filtration through the glomerulus, the substances and water in the primary urine undergo a reabsorption and secretion process in the tubules to reabsorb necessary substances and water into blood and excrete/discard unnecessary ones in the urine. Due to these processes, glomeruli and tubules are often known to be toxic targets for drugs.
It is, however, difficult to assess nephrotoxicity in vitro, since there are few in vitro methods to assess glomerulus toxicity due to the lack of appropriate primary cultured cells and cell lines (Pfaller and Gstraunthaler, 1998). Instead, in vivo toxicity studies using animals are useful for the evaluation of nephrotoxicity, along with urinalysis and pathological examinations. Since urinalysis is noninvasive and widely used in the clinic, it is easy to translate its results from nonclinical tests to clinical practice when abnormalities are detected in nonclinical toxicity studies. Generally, blood urea nitrogen (BUN) and serum creatinine (SCr) in blood chemistry, and total protein, albumin, and creatinine in urinalysis are used as indicators of nephrotoxicity (Dieterle et al., 2010; Ozer et al., 2010). Furthermore, several sensitive urinary biomarkers have been identified for the early detection and management of nephrotoxicity (Fuchs and Hewitt, 2011), such as kidney injury molecule 1 (Kim-1) and liver-type fatty acid binding protein (L-FABP) for renal tubular damage (Vaidya et al., 2010; Nan-ya et al., 2015), and neutrophil gelatinase-associated lipocalin (NGAL), cystatin C, and β2-microglobulin (β2M) for tubular and glomerular damage (Dieterle et al., 2010; Torregrosa et al., 2015; Prozialeck et al., 2016). When one of these biomarkers increases, it is easy to decide on the discontinuation of drugs to ensure patient safety. A pathological examination is also useful to identify the presence or absence of damage and toxicological target sites. Therefore, it is important to evaluate the potential for drug-induced nephrotoxicity in vivo. In particular, a urinalysis allows not only the detection and management of nephrotoxicity, but also the translation from animals to humans without invasive procedures.
K-975 appears to be useful for evaluating TEAD inhibition-mediated toxicity, including nephrotoxicity; however, the toxicity profile of K-975 remains to be evaluated (Kaneda et al., 2020). Verteporfin, approved by the US Food and Drug Administration as a photosensitizer to treat macular degeneration, is also considered to be useful for evaluating TEAD inhibition-mediated toxicity; however, verteporfin is known to have cytotoxic effects independent from TEAD inhibition as off-target effects (Messmer and Abel, 2001). Thus, TEAD inhibition-specific toxicity cannot be accurately assessed with verteporfin. Therefore, the evaluation of the toxicity profiles of TEAD inhibitors, their reversibility, and their management methods are needed for the development of TEAD inhibitors as anticancer agents. In particular, information on their long-term use is important for the clinical use of TEAD inhibitors and their combination with standard therapies, since anticancer drugs are generally administered with several repeated cycles of administration and withdrawal (Jänne et al., 2006).
In this study, we have evaluated the toxicity profile, including nephrotoxicity, of K-975 in rats and explored the management of K-975-mediated toxicity for the development of K-975 and other TEAD inhibitors as anticancer drugs.
MATERIAL AND METHODS
Test article
K-975 was synthesized by Kyowa Kirin Co., Ltd. (Tokyo, Japan).
Toxicity study in animals
K-975 was suspended in 0.5% methylcellulose 400 (0.5% MC or vehicle; FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) and repeatedly administered orally to male Crl:CD(SD) rats (The Jackson Laboratory Japan, Inc., Atsugi, Japan; n = 8 each) once a day at doses of 0 (0.5% MC alone), 30, 100 and 300 mg/kg of K-975 for 1 week. Five animals were sacrificed approximately 24 hr after the final seventh dose, the remaining 3 animals were autopsied at the end of a 2-week recovery period (represented as a “1-cycle study” in the manuscript). Similarly, K-975 suspended in 0.5% MC was administered orally to male rats (n = 10 each) at doses of 0 (0.5% MC alone), 100 and 300 mg/kg of K-975 for a 1-week administration period and 2-week recovery periods, and the cycle of administration and recovery periods was repeated 3 times (represented as a “3-cycle study” in the manuscript). Approximately 24 hr after the final (seventh) dose of the third cycle, the animals were divided into 2 groups so that the albumin index on urinalysis was evenly distributed between necropsy and recovery animals in the same dose level group, and five animals were autopsied. The remaining 5 animals were autopsied at the end of the 2-week recovery period in the third cycle.
On the day of necropsy, rats (fasting overnight only for the 1-cycle study) were anesthetized with isoflurane (Escaine, Mylan Pharmaceuticals, Morgantown, WV, USA), and blood samples for hematology and blood chemistry were collected in tubes with EDTA-2K for hematology or without anticoagulant for blood chemistry through the vena cava. Hematology was performed with XN-2000v (Sysmex Corporation, Kobe, Japan) and blood chemistry was performed with an H7180 (Hitachi High-Tech Corporation, Tokyo, Japan) and Epalyzer II Jr. (Helena Laboratories Co., Ltd., Saitama, Japan). Liver and kidney were collected for quantitative reverse transcription PCR (qRT-PCR) only in the 1-cycle study.
The following elements were evaluated: morbidity, mortality, clinical signs, body weight measurement, urinalysis, urine biomarkers (1-cycle study only), hematology (1-cycle study only), blood chemistry, necropsy, organ weights, histopathology, and gene expression analysis (1-cycle study only). Pathological examinations were conducted only for organs of the thoracic and abdominal cavity. All animal experiments were carried out according to the guidance of the institutional animal ethics committee of the testing facility (Kyowa Kirin Co., Ltd.).
Urinalysis and urine biomarker measurement
Urine was collected for approximately 16 hr (overnight) twice a week for the 1-cycle study with fasting only the day before necropsy, and once a week for the 3-cycle study without fasting at all points. Urinalysis was performed with a CLINITEK Advantus (Siemens Healthcare Diagnostics K.K., Tokyo, Japan), H7180 (Hitachi High-Tech Corporation), and PVA-EXII (A&T Corporation, Yokohama, Japan).
The urine biomarker was measured using the MILLIPLEX MAP Rat Kidney Toxicity Magnetic Bead Panel 1/2 (Merck KGaA, Darmstadt, Germany) for β2M, cystatin C, NGAL and Kim-1, and the Rat L-FABP ELISA Kit (CMIC HOLDINGS Co., Ltd., Tokyo, Japan) for L-FABP. L-FABP was undetectable (detection limit: 1.56 ng/mL) in most of the animals, and undetectable samples were represented as 0.
Albumin and urine biomarkers were corrected for urinary creatinine.
Immunohistochemistry (IHC)
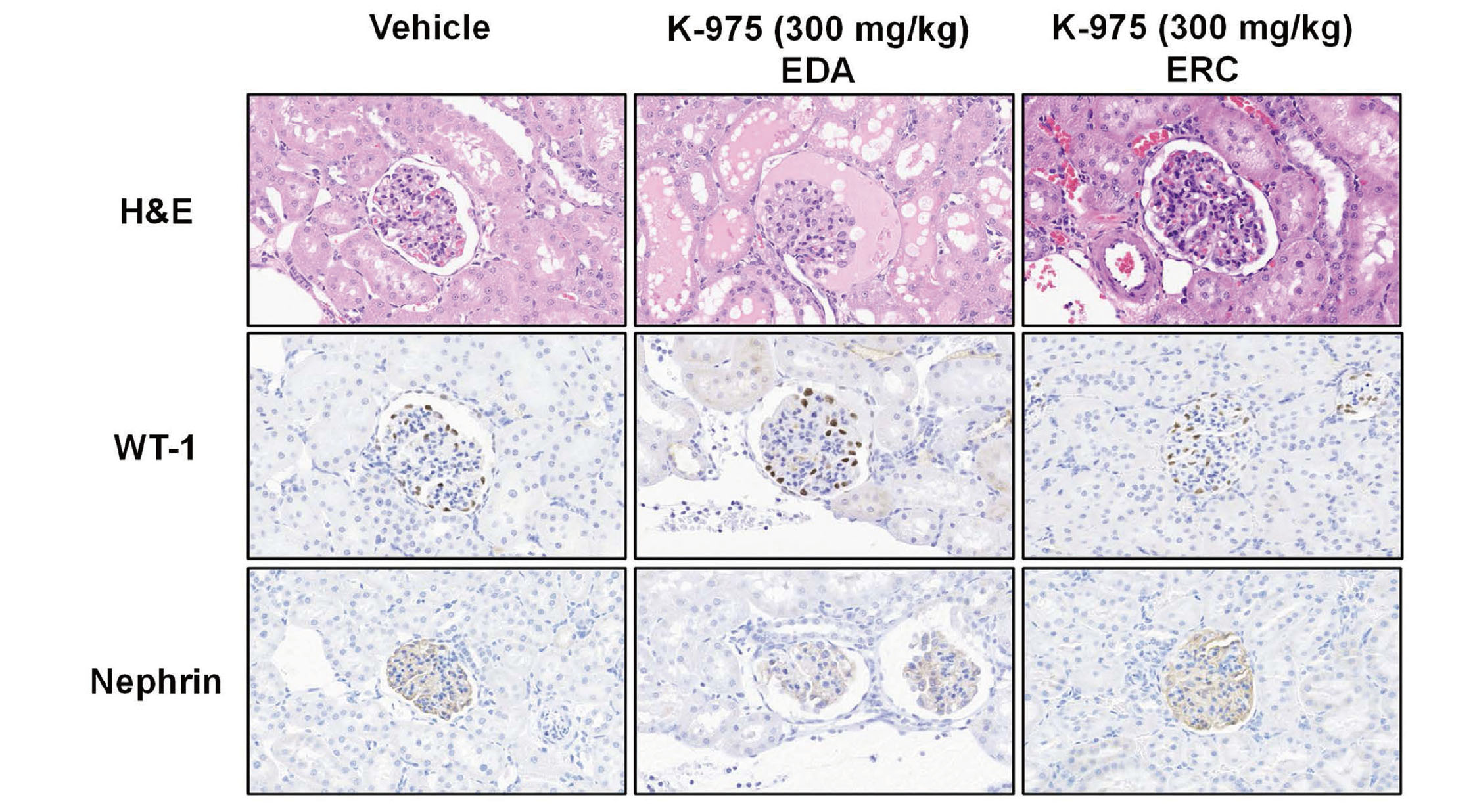
The kidneys of the rats in the 0 (0.5% MC) and 300 mg/kg groups of the 1-cycle study were collected, fixed in 10% (v/v) neutral buffered formalin solution, and embedded in paraffin. Kidney specimens were prepared from tissue and stained with antibodies against Nephrin (anti-Nephrin guinea pig polyclonal, serum; Cat No.: GP-N2, Lot No.: 606051-06, PROGEN Biotechnik GmbH, Heidelberg, Germany) and WT-1 (Anti WT1; clone: 6F-H2, Cat No.: MA1-46028, Lot No.: YA3804087, Invitrogen, Waltham, MA, USA) after heat retrieval with EnVision FLEX Target Retireval Solution (50x) Low pH for Nephrin and High pH for WT-1 (Agilent Technologies, Santa Clara, CA, USA).
Electron microscopy
A part of the kidney of the rats in the 0 (0.5% MC) and 300 mg/kg groups was fixed by immersion in 2.5% (v/v) glutaraldehyde. The kidney was sectioned into small pieces, washed/fixed with 0.1 mol/L phosphate buffer, osmium tetroxide solution, ethanol and epoxy mixture, and resin-embedded in blocks. Thick sections followed by ultrathin sections were prepared from the epoxy resin-embedded specimens. Ultrathin sections were electronically stained with uranyl acetate and lead citrate. Specimens were observed and representative photos were taken using a transmission electron microscope (JEM-1400Plus, JEOL Ltd., Tokyo, Japan). Thick section and subsequent examinations were performed at SHIN NIPPON BIOMEDICAL LABORATORIES, LTD. (Kagoshima, Japan). In the 1-cycle study, three animals were evaluated: one animal each in the 0 mg/kg and the 300 mg/kg groups at the end of the administration period, and the 300 mg/kg group at the end of the recovery period. In the 3-cycle study, four animals were evaluated: one animal each in the 0 mg/kg and the 300 mg/kg groups at the end of the administration period, and two animals in the 300 mg/kg group at the end of the recovery period.
Gene expression analysis
A gene expression analysis was performed only for the 1-cycle study. The liver and kidney were harvested from rats (n = 5 at the end of the administration period, n = 3 at the end of the recovery period), placed in tubes containing RNAprotect® Tissue Reagent (QIAGEN K. K., Tokyo, Japan), stored at 4°C overnight and frozen at -80°C the next day or later. On the day of RNA extraction, cryopreserved tissues were thawed at room temperature and RNA was extracted using the Maxwell RSC SimplyRNA Tissue Kit (Promega K.K., Tokyo, Japan) and cDNA preparation was performed using SuperScript IV VILO Master Mix (Invitrogen) according to the manufacturer’s protocol. For quantitative real-time PCR (qPCR), TaqMan™ Fast Universal PCR Master Mix (2X, no AmpErase™ UNG) and TaqMan™ Gene Expression Assays (Applied Biosystems, Waltham, MA, USA) were used according to the manufacturer’s protocols. Liver samples were evaluated in duplicate, and kidney samples were evaluated in singlicate. qPCR was performed with the 7500 Fast Real-Time PCR system (Applied Biosystems): Step 1, 95°C for 20 sec; step 2, 95°C for 3 sec; step 3, 60°C for 30 sec (steps 2 and 3 were repeated for 40 cycles). The evaluated genes and primer IDs were as follows: Gapdh (Rn01775763_g1), Hprt1 (Rn01527840_m1), Diaph3 (Rn01466145_m1), Ankrd1 (Rn00566329_m1), Cdh2 (Rn00580099_m1), Cdh1 (Rn00580109_m1), Ctgf (Rn00573960_g1), and Cyr61 (Rn01523136_g1). Gene expression levels in each sample were calculated using the ΔΔCt method normalized with internal controls (Gapdh and Hprt1).
Toxicokinetic analysis of K-975 in plasma
A toxicokinetic (TK) analysis was performed only for the 1-cycle study. Blood samples for TK analysis were collected through the tail vein (n = 3 each) using a restraint without fasting on Days 1 and 7 before dosing (pre, Day 7 only), 0.5, 1, 2, 4, 6, 8 and 24 hr after dosing. The samples were placed in tubes containing sodium heparin and plasma was obtained by centrifugation (approximately 5000 × g, 10 min, 4°C). Plasma K-975 concentrations were measured by liquid chromatography/tandem mass spectrometry (LC/MS/MS; LC, NexeraX2 series, Shimadzu, Kyoto, Japan; MS, QTRAP6500, AB SCIEX, Tokyo, Japan). The TK parameters were determined for K-975: the Cmax and the time to reach the Cmax (tmax) were obtained directly from the observed values. The area under the plasma concentration-time curve from time 0 to time t hr (AUC0-t) was calculated by the trapezoidal method from the observed values using Microsoft Excel for Office 365 (Microsoft, Seattle, WA, USA).
Statistical analysis
With the exception of the TK parameters, measured values were statistically analyzed using SAS version 9.4 or JMP version 17.0.0 (SAS Institute Inc., Cary, NC, USA). The Dunnett test or Steel test was performed for multiple comparisons between the corresponding vehicle group and each K-975 group. P values of < 0.05 were considered statistically significant.
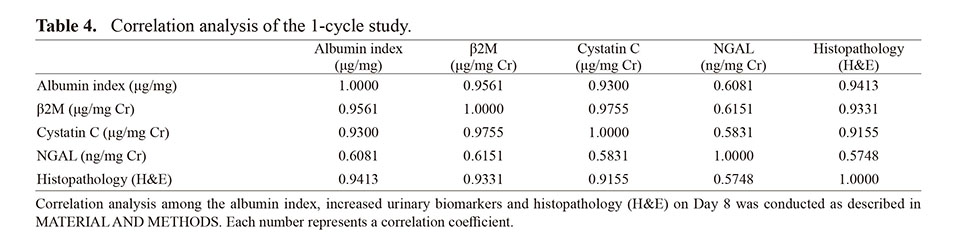
For the albumin index, urinary biomarkers, and histopathology (H&E) on Day 8, correlation analysis was conducted using JMP version 17.2.0 (SAS Institute Inc., Cary, NC, USA). Since there were multiple findings and grades in the pathology, each grade was converted to a numerical value (− = 0, ± = 1, and 1+ = 2), and the findings and grades for each animal were summed for correlation analysis.
RESULTS
K-975 exhibited high blood exposure and pharmacological effects in rats
To evaluate the toxicity profile of K-975, K-975 was administered orally to rats once a day for 1 week as the 1-cycle study (Supplementary Fig. 1). First, we conducted the TK evaluation of the concentration of K-975 in blood to confirm that K-975 was exposed to blood in rats. After a single dose of K-975, Cmax and AUC0-8hr on Day 1 increased almost linearly as the dose increased from 30 mg/kg to 300 mg/kg (Table 1). After repeated dosing for 7 days, Cmax and AUCs on Day 7 increased almost linearly as the dose increased from 30 mg/kg to 100 mg/kg. When the 100 and 300 mg/kg were compared on Day 7, Cmax was similar between the groups; however, AUC0-24hr was slightly higher and tmax was delayed in the 300 mg/kg group in comparison to the 100 mg/kg group (Table 1 and Supplementary Fig. 2), indicating that K-975 exposure was considered greater and longer at 300 mg/kg than at 100 mg/kg.
Table 1. Toxicokinetic analysis of the 1-cycle study.
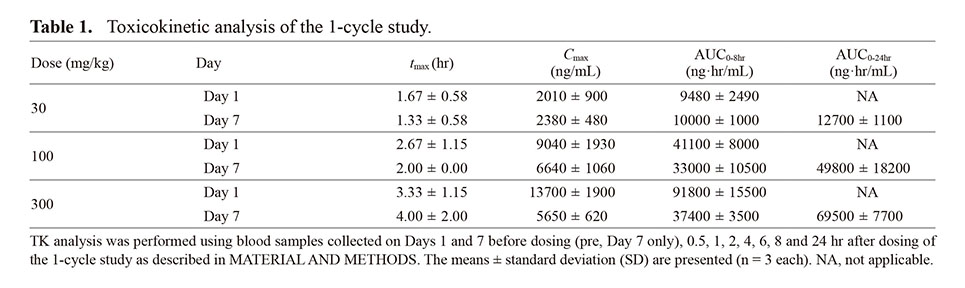
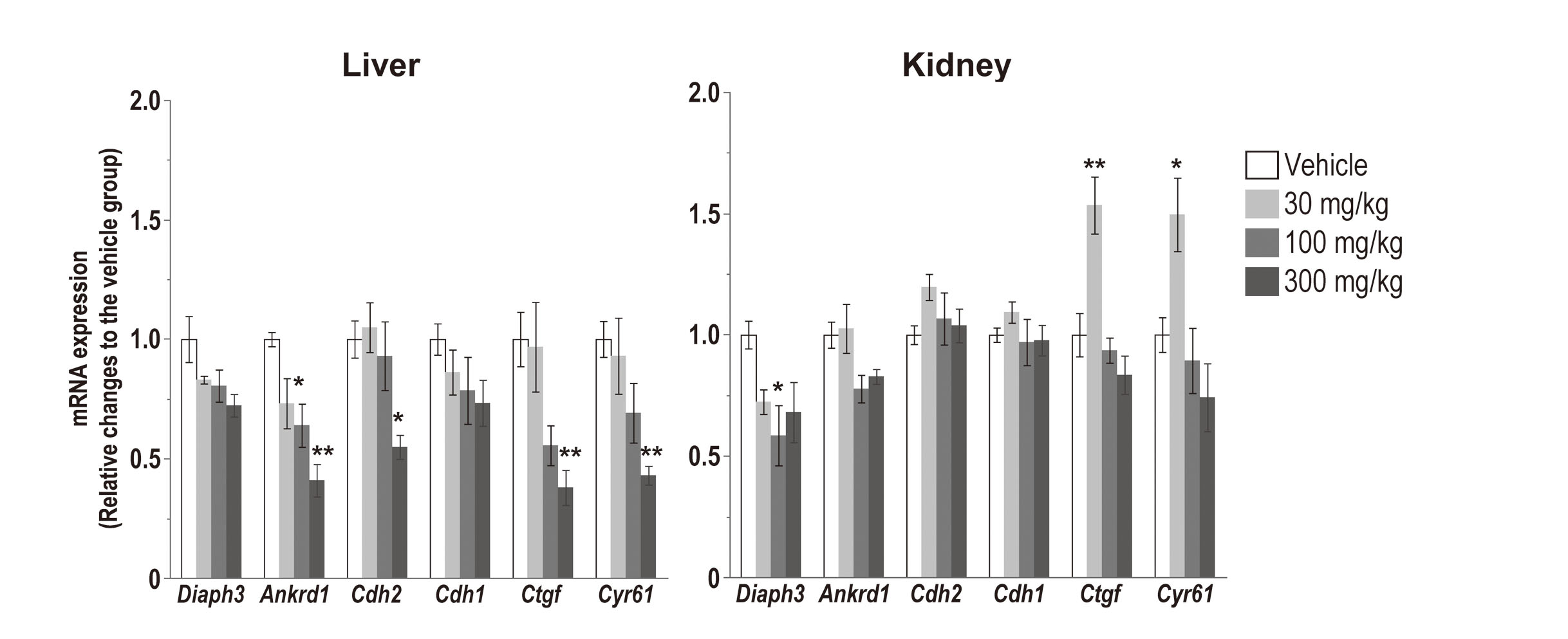
Next, we performed a gene expression analysis to confirm the pharmacological effects of K-975 in rats. The results of the liver and kidney analysis revealed that the expression of TEAD target genes such as Ankrd1, Cdh2, Ctgf, and Cyr61 was downregulated in the liver of the 300 mg/kg group (Fig. 1). These results proved that K-975 certainly inhibited the TEAD function and the expression of its downstream genes in rats. There were no apparent changes in the liver at doses of 100 mg/kg or less except for a decrease of Ankrd1 at 100 mg/kg. In the kidney, there were no statistically significant changes except for increases in Ctgf and Cyr61 at 30 mg/kg and a decrease in Diaph3 at 100 mg/kg, however, Diaph3, Ankrd1, Ctgf, and Cyr61 showed suppressive trends in the 300 mg/kg group.
K-975 induced nephrotoxicity in the 1-cycle study
We then evaluated the toxicity profiles of K-975. In the 1-cycle study, no K-975-related mortality, abnormalities in clinical signs (Supplementary Table 1), or change in body weight (Supplementary Table 2) were observed throughout the study period. Although crust formation and scarring in the 100 mg/kg group and reddish tear in the 300 mg/kg group were observed in one animal at each dose, they were not considered K-975-related changes because they were transient and occurred in only one case, and crust formation and scarring were observed only during the recovery period. Hematology (Supplementary Table 3) and blood chemistry (Table 2) showed no toxicological changes in all groups. These results indicated that the treatment with K-975 was well tolerated without life-threatening toxicity.
Table 2. Blood chemistry of the 1-cycle study.
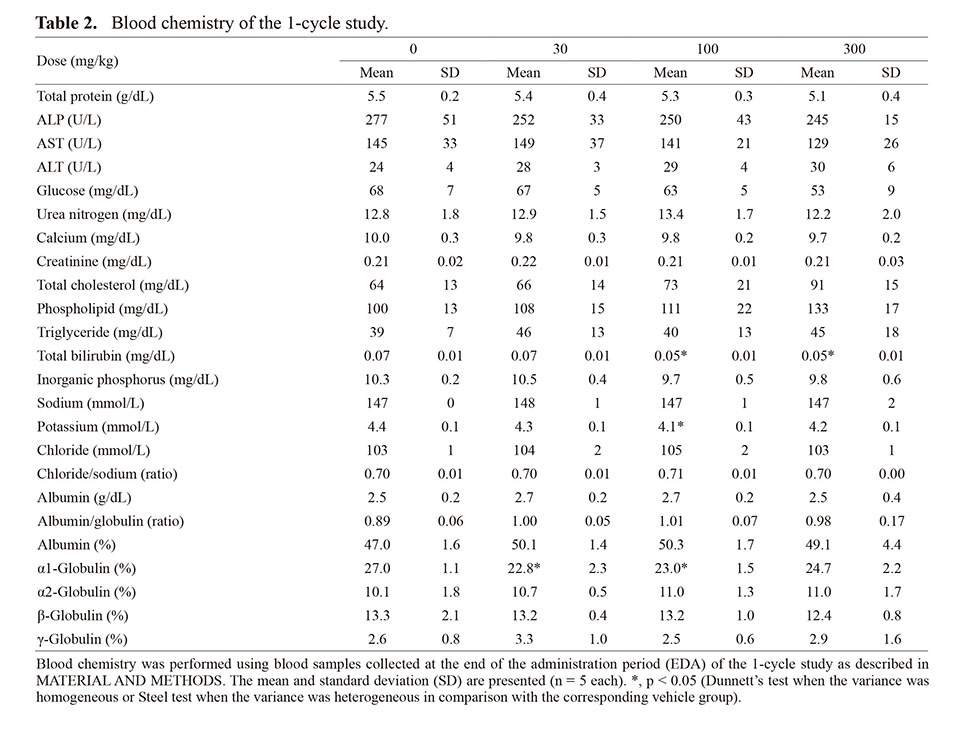
Urinalysis demonstrated that K-975 treatment showed statistically significant increases in urinary albumin concentrations and albumin indices (urinary albumin/urinary creatinine) at 100 and 300 mg/kg and an increase in urinary total protein concentrations at 300 mg/kg (Fig. 2, Supplementary Fig. 3). A decrease in urinary creatinine concentration was also observed at 300 mg/kg, however, there were no obvious changes in the total amount of excreted creatine and creatinine clearance. There were no other abnormalities in the urinalysis results in all groups (Fig. 2, Supplementary Table 4), including urine volumes, indicating that K-975 treatment induced the increased excretion of total protein and albumin rather than urine condensation. However, there were no obvious changes in serum total protein or albumin concentrations, as described above (Table 2). Collectively, the urinalysis results indicated that 1 week of K-975 administration caused proteinuria but did not affect the kidney function or general conditions in rats.
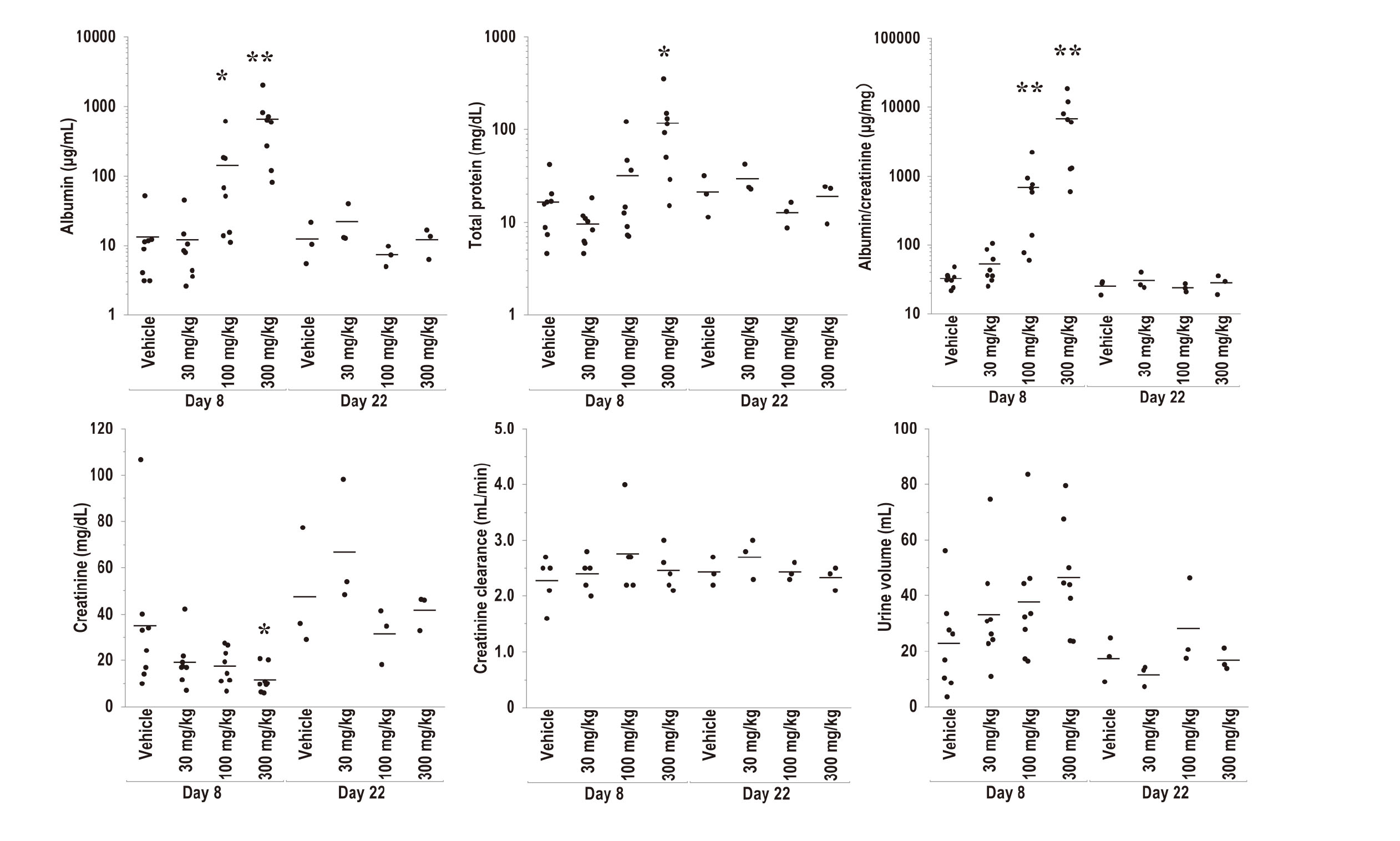
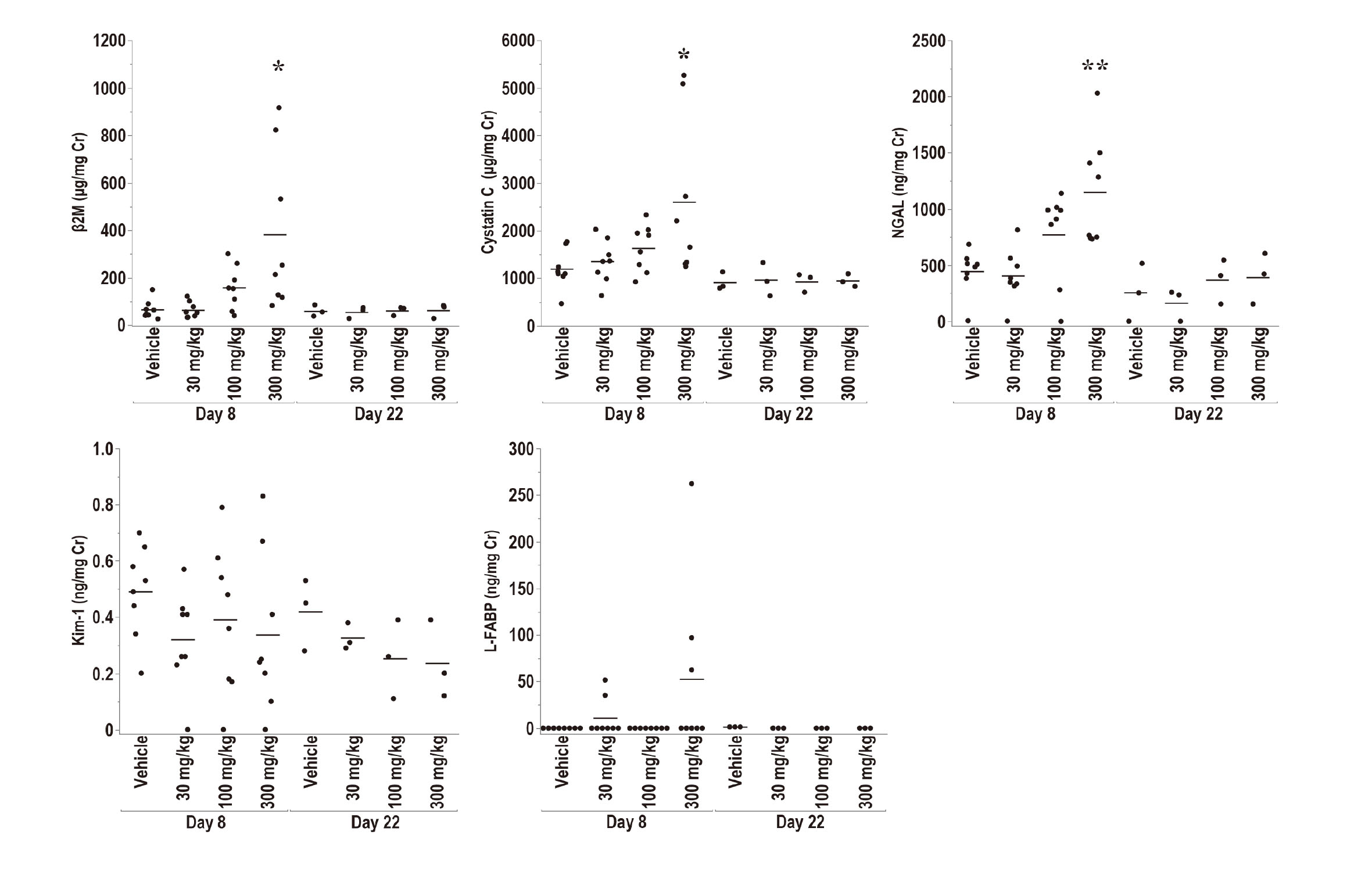
Since the urinalysis results demonstrated K-975-induced nephrotoxicity, the following urinary biomarkers were measured to investigate whether nephrotoxicity could be detected more sensitively and earlier: β2M, cystatin C, and NGAL for glomerulus and tubule toxicity, and Kim-1 and L-FABP for tubule toxicity. The measurement results showed statistically significant increases in β2M, cystatin C and NGAL at 300 mg/kg on Day 8 in comparison to the vehicle group. However, no obvious changes were observed in Kim-1 and L-FABP (Fig. 3, Supplementary Fig. 4). These findings indicate that K-975 affects the glomeruli rather than the tubules and that K-975-induced nephrotoxicity could be detected with three urinary markers suggesting glomerular toxicity.
K-975 caused the loss of the characteristic structure of the podocyte
A pathological examination revealed the main characteristic effects of K-975 on the kidneys. At the end of the administration period, macroscopical necropsy showed that the discolored kidney was observed bilaterally in the 300 mg/kg group (Table 3). A histopathological examination of the kidney demonstrated hyaline casts, hyalinization in the glomerular Bowman's capsule, and mitosis and single cell necrosis in glomerular cells in the 300 mg/kg group, although these changes were slight with few obvious injury lesions (Table 3). Furthermore, centrilobular hypertrophy of hepatocytes was observed in the 300 mg/kg group, corresponding to a statistically significant increase in organ weight (Supplementary Table 5). Degeneration/necrosis was also observed in Brunner’s gland of the small intestine in the 300 mg/kg group. No other abnormalities associated with the administration of K-975 were observed in the organs/tissues that we examined. These results indicate that K-975 mainly affects the kidneys, with only minor changes in the liver and small intestine.
Table 3. Necropsy and histopathology of the 1-cycle study.
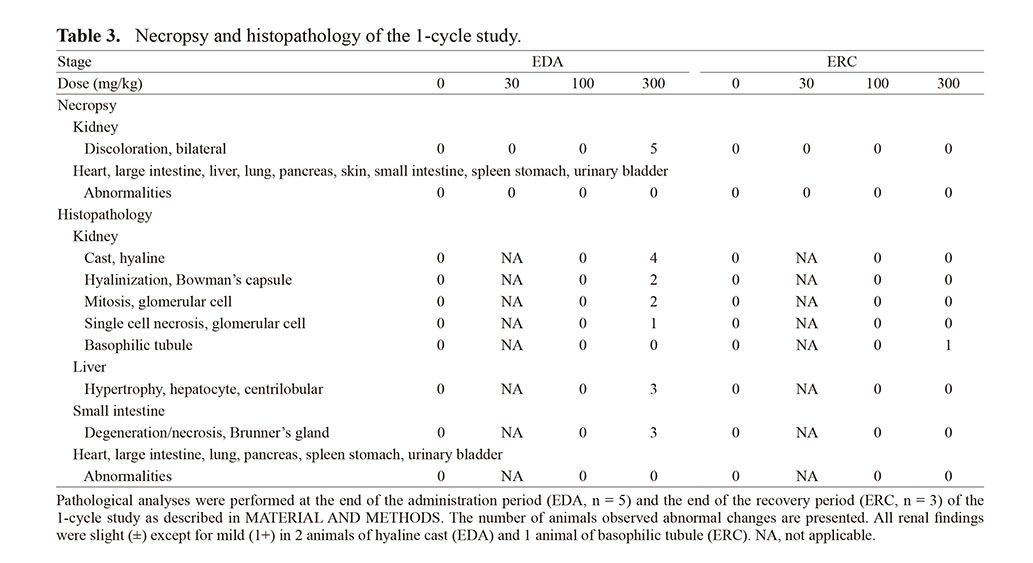
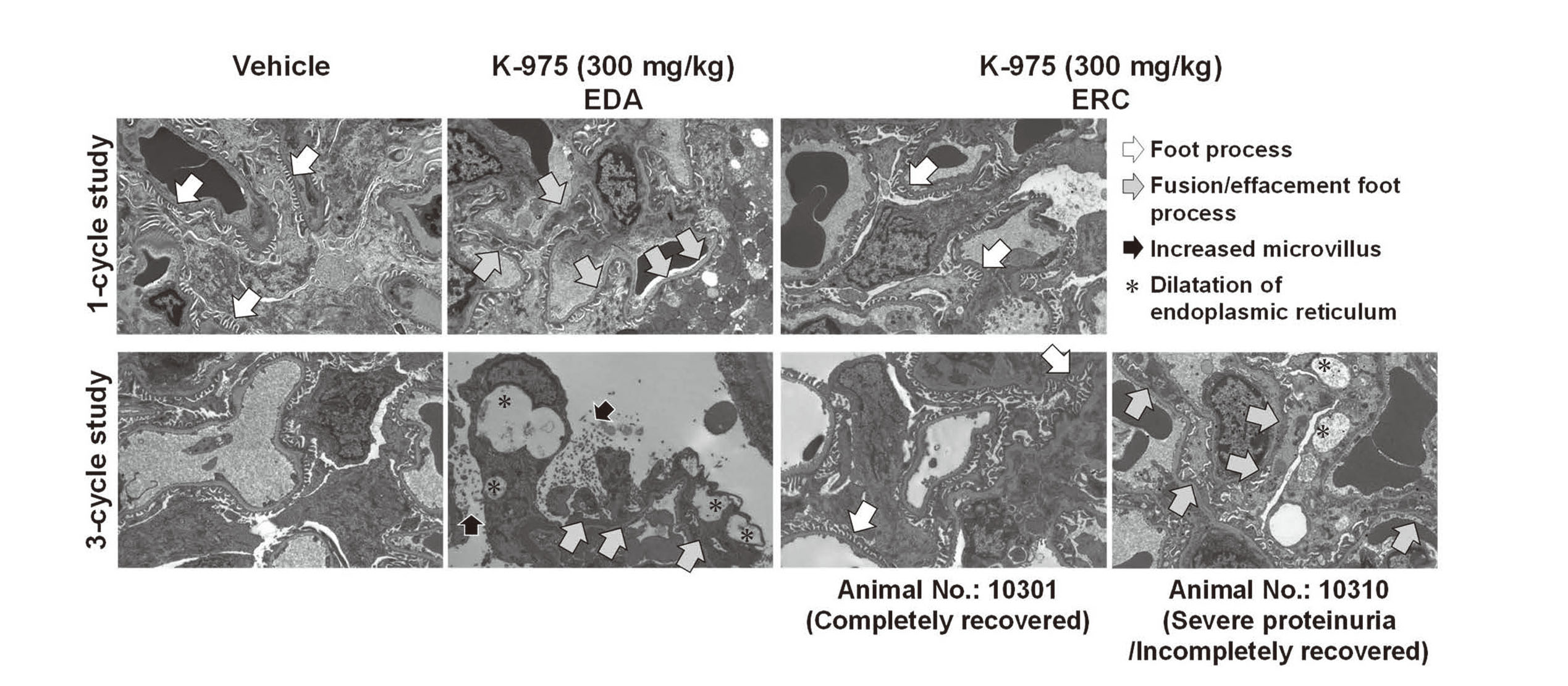
Although the results of the urinalysis and urinary biomarker measurement indicated the effect of K-975 on the glomerulus, there were only slight changes with few obvious injury lesions on histopathological examination with H&E staining (Fig. 4). Thus, we performed IHC and electron microscopy evaluations of the kidney, considering possible effects on the glomeruli. In the IHC evaluations, WT-1 and Nephrin, a nuclear marker of podocytes and a marker of podocyte foot processes, respectively, were stained to assess the effect of K-975 on the glomeruli in detail (Fig. 4). The results showed that K-975 treatment at 300 mg/kg decreased Nephrin staining. On the other hand, there was no change in the number of WT-1 positive cells in the glomerulus. Furthermore, electron microscopy of the glomerulus revealed that treatment with K-975 at 300 mg/kg caused fusion/effacement of foot processes in podocytes (Fig. 5). These results indicate that K-975 induces proteinuria by altering the characteristic structure of podocytes rather than by injuring glomerular podocytes.
To confirm the relationship among the changes caused by K-975, correlation analyses were conducted using individual values of the albumin index, increased three urinary biomarkers (β2M, cystatin C, and NGAL), and histopathology (H&E) on Day 8. The results showed that these changes, including albumin index, were positively correlated (Table 4), indicating that the nephrotoxicity of K-975 was based on the kidney glomerular changes.
Table 4. Correlation analysis of the 1-cycle study.
 K-975-induced nephrotoxicity showed reversibility after a recovery period in the 1-cycle study
K-975-induced nephrotoxicity showed reversibility after a recovery period in the 1-cycle study
Next, we evaluated the reversibility of K-975-induced toxicities with a 2-week recovery period after a 1-week administration period. At the end of the recovery period, there were no abnormalities among all of the items examined in each of the group. In other words, the K-975-induced nephrotoxicity observed during the administration period, such as severe proteinuria (urinalysis, Fig. 2), increased urinary biomarkers (Fig. 3), and podocytes foot process effacement (IHC and electron microscopy, Fig. 4 and 5), completely recovered after a 2-week recovery period. In addition, K-975-mediated changes, including increased liver weight and degeneration/necrosis in Brunner's gland of the small intestine, recovered (Table 3, Supplementary Table 5). These results indicate that the toxicity of K-975, including severe proteinuria, is reversible.
K-975-induced nephrotoxicity was reversible in the 3-cycle study
The results of the 1-cycle study revealed that K-975-induced nephrotoxicity was reversible. However, it was not clear whether the changes were reversible even after long-term treatment with K-975. Therefore, we conducted a longer-term study of K-975 with 3 cycles of 1-week dosing and 2-week recovery (Supplementary Fig. 1) to evaluate whether nephrotoxicity was reversible and not exacerbated by longer-term treatment.
Since the albumin index was identified as a sensitive marker for K-975-induced nephrotoxicity in the 1-cycle study, the albumin index was measured as a nephrotoxicity indicator in the 3-cycle study (Fig. 6A). When comparing the albumin index at the end of dosing in each cycle, the degree of proteinuria worsened in the 100 mg/kg group, but the changes in the index were almost the same in the 300 mg/kg group. In both the 100 and 300 mg/kg groups, although proteinuria showed a trend toward recovery at the end of the recovery period in the 1-cycle study, it did not fully recover at the end of the second or third recovery period in the 3-cycle study (Fig. 6A). However, the individual albumin indices showed that only one animal each in the 100 and 300 mg/kg groups (animal Nos. 10207 and 10310), which had the highest albumin index in each group, showed slow and incomplete recovery after the second or later recovery period, while the other animals in both groups had low values and fully recovered, even at the end of the third recovery period (Fig. 6B, 6C). In the two animals that showed a slow recovery, the albumin index showed an upward trend with repeated dosing cycles, even at the end of the recovery period of each cycle. These results indicate that K-975-induced nephrotoxicity could be reversible, even after 3 repeated cycles of 1-week administration and 2-week recovery periods, but that resumption of K-975 dosing with incomplete recovery could exacerbate nephrotoxicity.
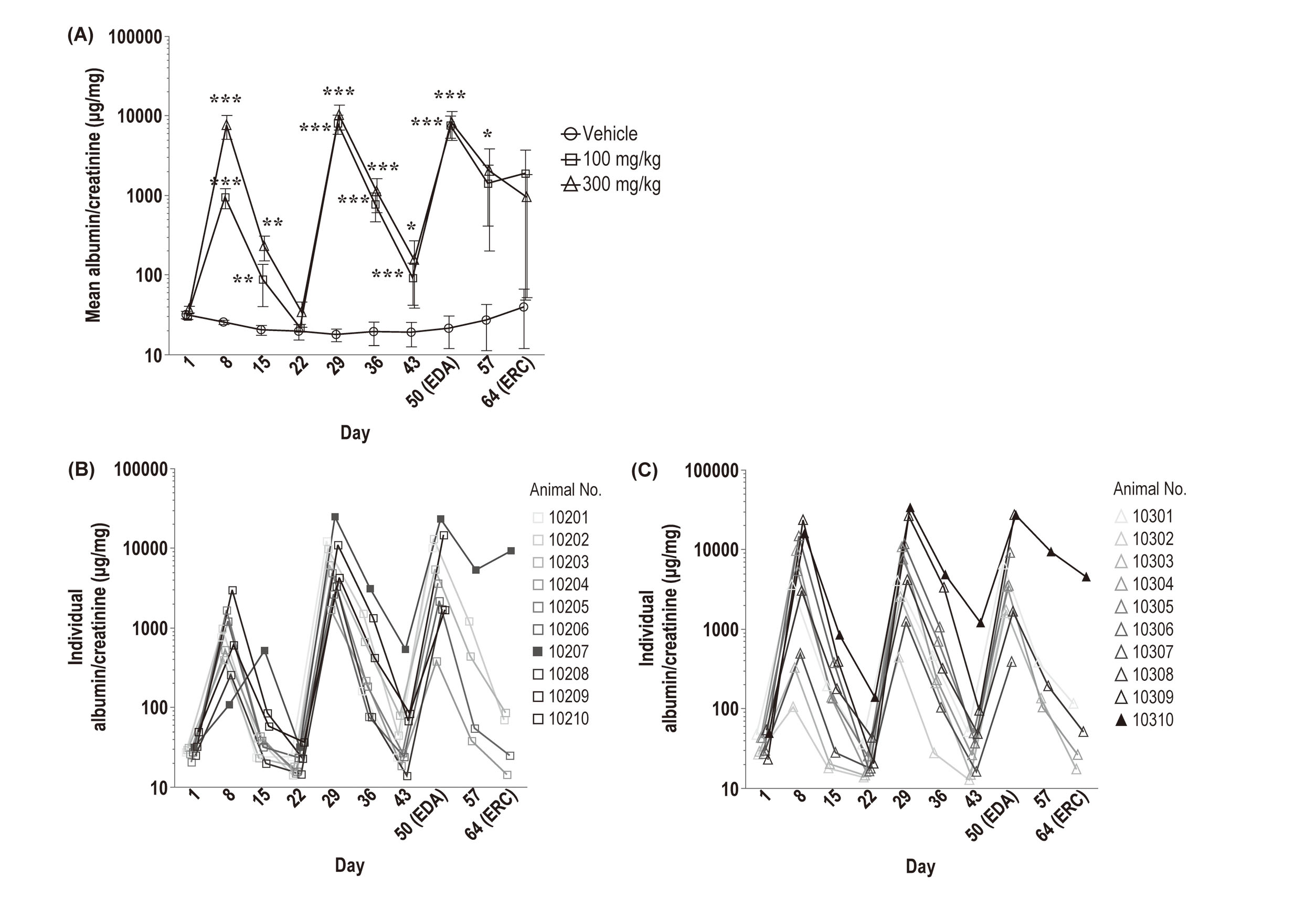
Two animals with high albumin indices showed a slow and incomplete recovery, indicating the exacerbation of proteinuria. Therefore, we performed pathological examinations of the kidneys to examine what changes occurred in these animals with 3-cycle of administration and recovery. The histopathologic examination revealed stronger nephrotoxic changes in these two animals than other animals: glomerular atrophy in animal No. 10310 at 300 mg/kg, and basophilic tubule, hyaline cast, and interstitial fibrosis in animal Nos. 10207 at 100 mg/kg and 10310 at 300 mg/kg, where severe proteinuria occurred (Supplementary Table 6). Furthermore, electron microscopy revealed that the fusion/effacement of the podocyte foot processes did not fully recover at the end of the third recovery period in animal No. 10310 (Fig. 5). These results indicate that K-975 treatment could cause tissue injuries with the exacerbation of proteinuria by resuming K-975 treatment before proteinuria has fully recovered, possibly resulting in non-reversibility.
DISCUSSION
In this study, we evaluated the toxicity profiles of K-975, a novel TEAD inhibitor, in rats by repeated oral administration for the 1- or 3-cycles of 1-week administration and 2-week recovery periods. The results of the present study have revealed that the main toxicological concern of K-975 is nephrotoxicity, but it is reversible and monitorable by the albumin index.
After 1-week treatment with K-975, nephrotoxicity, hepatocellular hypertrophy, and degeneration/necrosis were observed in Brunner’s gland of the small intestine (Fig. 2 and Table 3). The liver changes caused by K-975 were not considered to be toxic due to the absence of abnormal blood liver enzymes or injurious lesions (Tables 2 and 3). Furthermore, it was speculated that the change in the small intestine was independent of TEAD inhibition and a direct effect of the K-975 dosing formulation since double-deficient Yap/Taz mice showed no abnormalities in the homeostasis and architecture of small intestine (Azzolin et al., 2014). In addition, it was judged to have a low clinical extrapolability because the drugs would be taken in tablet form in the clinic. Therefore, we consider nephrotoxicity to be the main toxicological concern of K-975.
K-975 showed severe proteinuria with fusion/effacement of podocyte foot processes after a 1-week administration (Fig. 2, 3, and 5). Podocyte foot processes play an important role as a filter from blood into urine, especially in preventing the leakage of macromolecules in plasma such as serum albumin (Pavenstädt et al., 2003; Reiser and Altintas, 2016). On the contrary, the loss of foot processes leads to the leakage of serum proteins into the urine, proteinuria, and has been observed in many proteinuric renal diseases (Mundel and Reiser, 2010). Furthermore, proteinuria has been reported in podocyte-specific Yap cKO mice (Schwartzman et al., 2016). Therefore, K-975-induced proteinuria is considered to occur due to loss of podocyte foot processes in the glomerulus.
The molecular mechanism of the fusion/effacement of the podocyte foot process by inhibition of TEAD remains to be elucidated, but it is possible that K-975 may inhibit the maintenance of the TEAD-mediated cytoskeleton in podocytes. Podocyte foot processes expresses several cytoskeletal molecules (i.e. actin) to maintain their characteristic structure (Reiser and Altintas, 2016). The downstream effectors of the Hippo pathway are also known to contribute to cytoskeletal maintenance: TAZ is known to transcriptionally activate the expression of synaptopodin, zonula occludens-1 (ZO-1) and zonula occludens-2 (ZO-2) in a TEAD-dependent manner (Chen et al., 2022). Synaptopodin, an actin cytoskeletal protein, is expressed in podocyte foot processes and plays an important role in maintaining the integrity of the actin backbone (Mundel et al., 1997). Although Synaptopodin KO mice do not exhibit phenotypes, the downregulation of its expression is also reported in nephrotic syndrome (Srivastava et al., 2001). ZO-1 and ZO-2, tight junction proteins, are expressed in mature podocytes and are required for cell-cell interaction (Scott and Quaggin, 2015). Although glomerulus-specific ZO-2 deficient mice also do not exhibit a phenotype, ZO-1 deficient mice develop severe proteinuria and double-deficient ZO-1/ZO-2 mice is known to further deteriorate the podocyte function (Itoh et al., 2014, 2018). Additionally, the Hippo pathway has been reported to be involved in the negative regulation of various cytoskeleton molecules, such as F-actin (Matsui and Lai, 2013) and integrin subunits (Nardone et al., 2017) by inhibiting polymerization and protein-protein interaction or reducing gene expression levels. Thus, not only the molecules mentioned here, but also other downstream molecules of the Hippo pathway are possibly involved in maintaining the glomerular cytoskeleton and homeostasis. Taken together, TEAD inhibition by K-975 is suggested to disrupt podocyte cytoskeletons by inhibiting cytoskeletal molecules downstream of the Hippo pathway.
Furthermore, glomerular homeostasis and function were restored through cytoskeleton regeneration after drug withdrawal, since K-975 only inhibited podocyte cytoskeleton maintenance and showed little cytotoxicity to podocytes. Although K-975 induced severe proteinuria, all changes, including proteinuria, had recovered after a 2-week recovery period (Fig. 2 to 5 and Table 3). This reversible nephrotoxicity by K-975 was different from the nephrotoxicity of well-known nephrotoxicants targeting the glomerulus, such as puromycin and adriamycin. Puromycin is used to induce a model of glomerular proteinuria in rats, and causes significant changes in podocytes, including loss of foot processes and decreased actin cytoskeleton levels. Although puromycin-induced nephrotoxicity was reported to be reversible 20 days after a single dose, the number of podocytes decreased even after a single dose of puromycin and decreased further after three repeated doses every 30 days (Kim et al., 2001). Additionally, a single dose of adriamycin caused proteinuria that lasted for more than 7 weeks (Wang et al., 2000). Furthermore, the exacerbation of glomerulosclerosis and a decreasing glomerular count over time were also observed. These results indicate that puromycin- and adriamycin-induced proteinuria is a pathologically and functionally progressive nephrotoxicity and that K-975 is safer than these well-known nephrotoxicants, as its nephrotoxicity showed reversibility.
The mechanistic difference between K-975 and puromycin/adriamycin is unclear; however, one possible reason for the difference is the cytotoxic effect of these compounds. Puromycin and adriamycin are known to produce reactive oxygen species, and podocyte injury by puromycin can be reduced by antioxidants (Thakur et al., 1988; Ricardo et al., 1994). These reports suggest that cytotoxicity causes nephrotoxicity. In general, podocytes are not regenerated, so renal damage caused by these compounds could be difficult to recover. On the other hand, despite severe proteinuria, K-975 showed no obvious injurious changes on histopathological examination and no change in glomerular WT-1 at the end of the administration period (Fig. 4 and Table 3). These results indicate that K-975 has no cytotoxic effects. As described above, K-975-induced nephrotoxicity was caused by podocyte cytoskeleton alteration by inhibiting cytoskeletal molecules downstream of the Hippo pathway. Furthermore, glomerular homeostasis and function could be restored through cytoskeleton regeneration after drug withdrawal without cytotoxicity to podocytes. Therefore, K-975-induced reversible nephrotoxicity is suggested to be caused not by damage to tissues or cells but by cytoskeleton alterations, which could be why K-975 differs from puromycin and adriamycin.
To identify safety biomarkers that allow for the early detection and management of K-975-induced nephrotoxicity, we measured several urinary biomarkers and found that levels of β2M, cystatin C, and NGAL, biomarkers for glomerular and tubular damage (Dieterle et al., 2010; Torregrosa et al., 2015; Prozialeck et al., 2016), increased in the highest dose group (300 mg/kg), while Kim-1 and L-FABP, biomarkers for tubular damage, did not change (Fig. 3). The three elevated biomarkers are present in the blood, filtered by the glomeruli, and reabsorbed in the tubules under normal conditions. During glomerular injury, the reabsorption of these urinary biomarkers in the tubules is inhibited by urinary proteins such as albumin, resulting in elevated urinary biomarkers (Thielemans et al., 1994). In addition, NGAL is known to be expressed in the renal tubules during acute kidney injury (Mårtensson and Bellomo, 2014). In the present study, K-975 showed proteinuria in urinalysis (Fig. 2) and few apparent renal injuries in the kidney (Table 3), suggesting that all three biomarkers were inhibited by proteinuria from being reabsorbed in the renal tubules. These findings and mechanistic interpretations suggest that urinary biomarkers of glomerular toxicity are helpful in detecting K-975-induced nephrotoxicity.
The results of urinalysis and urinary biomarker analyses demonstrate that urinary albumin concentration and albumin index in urine were the most sensitive markers, increasing from a dose of 100 mg/kg (Fig. 2 and 3). The albumin index is considered very useful for the management of K-975-induced nephrotoxicity as a translational safety biomarker from nonclinical studies to clinical trials, as the urinary albumin concentration and albumin index can be easily measured by routine urinalysis, and the albumin index is used as an indicator of proteinuria in clinical practice. However, in humans, proteinuria is diagnosed when the albumin index is 30 mg/g or greater, and the mean (interquartile range) albumin index is 555 mg/g (159-1556 mg/g) even in patients with stage 4 CKD (Wu et al., 2012), while the albumin index of rats treated with K-975 at 300 mg/kg increased to approximately 7000-8000 mg/g. Therefore, monitoring the degree of changes in the albumin index will be important for determining discontinuation according to established criteria (e.g., 30 mg/g) in humans.
The reversibility of K-975-induced nephrotoxicity was generally similar after 3 cycles of administration and recovery periods (Fig. 6). However, the tendency for a slow recovery and incomplete recoverability were observed in one animal with the highest albumin index in each dosing group (animal Nos. 10207 and 10310 at 100 and 300 mg/kg, respectively; Fig. 6B and 6C). In these two animals, proteinuria had not recovered fully at the end of each recovery period before the resumption of K-975 treatment. In the histopathological examination of these animals, the glomerulus showed glomerular atrophy and/or stronger interstitial fibrosis than in other animals (Supplementary Table 6). Fibrosis was the result of chronic tubulointerstitial damage following glomerular injury and is generally an irreversible change, which could be related to poor recovery of proteinuria. In addition, diphtheria toxin-induced podocyte depletion of up to 20% was reported to be restored to a normal structure, while podocyte depletion of more than 20% was associated with irreversible glomerular damage, leading to progressive renal failure (Wharram et al., 2005). From our results and these reports, K-975-induced nephrotoxicity could also be reversible, even after 3 cycles of administration and recovery periods; however, the severe proteinuria induced by continuous treatment with K-975 can cause irreversible podocytopenia and glomerular degeneration, which is a risk factor for kidney failure. Therefore, for the clinical use of K-975, it seems important to monitor and manage proteinuria using the albumin index to ensure patient safety.
TK analysis revealed the high blood exposure of K-975. Cmax and AUCs increased almost linearly as the dose increased from 30 mg/kg to 100 mg/kg. At the same time, non-linear Cmax and AUC0-24hr were observed between 100 and 300 mg/kg groups (Table 1 and Supplementary Fig. 2). These results could be due to the relationship between the Hippo pathway and drug-metabolizing enzymes. For example, YAP has been reported to interact with the nuclear receptor PXR, which regulates the expression of various drug-metabolizing enzymes, to inhibit the PXR-mediated induction of drug-metabolizing enzymes (Abe et al., 2019). Conversely, in this study, it was speculated that TEAD inhibition by K-975 might have canceled PXR inhibition by YAP, resulting in the induction of the drug-metabolizing enzymes. Furthermore, gene expression analysis supported this hypothesis because K-975-mediated TEAD inhibition was observed in the livers of rats in 100 and 300 mg/kg groups.
Despite the high blood exposure of K-975 (Table 1) and morphological changes in the liver and kidney (Fig. 2 and Table 3), the evaluation of the gene expression revealed statistically significant changes only in the liver and non-significant suppressive trends in the kidney (Fig. 1). These results are probably attributable to the heterogeneity and complexity of the organ. The liver is a relatively homogeneous tissue composed mainly of hepatocytes, which might have made it easy to detect the TEAD inhibition effect of K-975 while the kidney is composed of various types of cells. Furthermore, K-975-induced nephrotoxicity manifested as proteinuria by the glomerular foot process fusion/effacement with increased urinary biomarkers of glomerular damage (Fig. 2, 3, and 5), suggesting that K-975 does not act on the kidney as a whole, but only on the glomeruli, particularly on the podocytes. In addition, this kidney glomerulus-specific effect of K-975 was supported by the correlation analysis of albumin index with urinary biomarkers and histopathology (Table 4). Collectively, the finding that the effects of TEAD inhibition were particularly pronounced only in glomerular podocytes may explain why only slight gene expression changes were observed in the kidney. It should be noted that hepatocyte hypertrophy and increased liver weight without abnormal blood liver enzymes (e.g., AST and ALT) that were observed with the administration of K-975 (Table 2 and 3 and Supplementary Table 5) have also been reported in Yap or Taz cKO mice (Lu et al., 2018), indicating that K-975 actually inhibited TEAD and the expression of its downstream genes in the liver of rats in this study.
In addition to the kidney, liver, and small intestine, K-975 did not affect other organs, such as the heart and lung (Table 3), which is in contrast to previous reports with Yap or Taz cKO mice (Makita et al., 2008; Xin et al., 2013). One of the reasons for the differences between the phenotypes of cKO mice and the toxicity profiles observed with inhibitor administration in rats may be the different roles of TEAD in developmental stages/juvenile animals and adult animals. The Hippo pathway and its downstream effectors are important for development, cell proliferation, and organ growth in various organs; however, outside of the glomerulus, the Hippo pathway and its downstream effectors might not be necessary in adult animals. In drug discovery, information about KO/cKO mice is very important for predicting on-target toxicity by inhibition of target molecules; however, not all toxicological concerns from KO/cKO mice could be observed in the evaluation of the nonclinical toxicity of the inhibitors, as shown in the present study. Therefore, it is important to evaluate the actual toxicity of inhibitors rather than to rely on the results of KO/cKO mice, especially for drug development. Therefore, the present results provide crucial insights for the drug development of TEAD inhibitors, including K-975.
In conclusion, we evaluated the toxicity of K-975 in rats and revealed that K-975 induced glomerular podocyte foot process fusion/effacement, resulting in proteinuria, but there were few other organ toxicities. Our results suggest that K-975-induced nephrotoxicity is reversible after an appropriate recovery period and that the albumin index is a sensitive biomarker of K-975-induced proteinuria, indicating that this nephrotoxicity is monitorable by monitoring the albumin index. We believe that our findings will provide valuable information for the development of K-975 and other TEAD inhibitors as anticancer drugs.
ACKNOWLEDGMENTS
The authors deeply appreciate Masatomo Kajihara, Natsuko Iwasaki, Maiko Kagebayashi, Ayako Miwa, Rie Hirano, Toyoko Kashiwagi, Yoko Hasegawa for their technical assistance.
Conflict of interest
H.O., T. U., Y. I., Y. S., T. A., and K. N. are employees of Kyowa Kirin, Co., Ltd.
REFERENCES
- Abe, T., Shizu, R., Sasaki, T., Shimizu, Y., Hosaka, T., Kodama, S., Matsuzawa, A. and Yoshinari, K. (2019): Functional interaction between pregnane X receptor and yes-associated protein in xenobiotic-dependent liver hypertrophy and drug metabolism. J. Pharmacol. Exp. Ther., 371, 590-601.
- Azzolin, L., Panciera, T., Soligo, S., Enzo, E., Bicciato, S., Dupont, S., Bresolin, S., Frasson, C., Basso, G., Guzzardo, V., Fassina, A., Cordenonsi, M. and Piccolo, S. (2014): YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell, 158, 157-170. Elsevier.
- Camargo, F.D., Gokhale, S., Johnnidis, J.B., Fu, D., Bell, G.W., Jaenisch, R. and Brummelkamp, T.R. (2007): YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol., 17, 2054-2060.
- Chan, P., Han, X., Zheng, B., DeRan, M., Yu, J., Jarugumilli, G.K., Deng, H., Pan, D., Luo, X. and Wu, X. (2016): Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol., 12, 282-289.
- Chen, J., Wang, X., He, Q. and Harris, R.C. (2022): TAZ is important for maintenance of the integrity of podocytes. Am. J. Physiol. Renal Physiol., 322, F419-F428.
- Cunningham, R. and Hansen, C.G. (2022): The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. (Lond.), 136, 197-222.
- Dieterle, F., Perentes, E., Cordier, A., Roth, D.R., Verdes, P., Grenet, O., Pantano, S., Moulin, P., Wahl, D., Mahl, A., End, P., Staedtler, F., Legay, F., Carl, K., Laurie, D., Chibout, S.D., Vonderscher, J. and Maurer, G. (2010): Urinary clusterin, cystatin C, β2-microglobulin and total protein as markers to detect drug-induced kidney injury. Nat. Biotechnol., 28, 463-469.
- Dupont, S., Morsut, L., Aragona, M., Enzo, E., Giulitti, S., Cordenonsi, M., Zanconato, F., Le Digabel, J., Forcato, M., Bicciato, S., Elvassore, N. and Piccolo, S. (2011): Role of YAP/TAZ in mechanotransduction. Nature, 474, 179-183.
- Fuchs, T.C. and Hewitt, P. (2011): Biomarkers for drug-induced renal damage and nephrotoxicity-an overview for applied toxicology. AAPS J., 13, 615-631.
- Fujii, M., Toyoda, T., Nakanishi, H., Yatabe, Y., Sato, A., Matsudaira, Y., Ito, H., Murakami, H., Kondo, Y., Kondo, E., Hida, T., Tsujimura, T., Osada, H. and Sekido, Y. (2012): TGF-β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med., 209, 479-494.
- Haskins, J.W., Nguyen, D.X. and Stern, D.F. (2014): Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal., 7, ra116.
- Hossain, Z., Ali, S.M., Ko, H.L., Xu, J., Ng, C.P., Guo, K., Qi, Z., Ponniah, S., Hong, W. and Hunziker, W. (2007): Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc. Natl. Acad. Sci. USA, 104, 1631-1636.
- Itoh, M., Nakadate, K., Horibata, Y., Matsusaka, T., Xu, J., Hunziker, W. and Sugimoto, H. (2014): The structural and functional organization of the podocyte filtration slits is regulated by Tjp1/ZO-1. PLoS One, 9, e106621.
- Itoh, M., Nakadate, K., Matsusaka, T., Hunziker, W. and Sugimoto, H. (2018): Effects of the differential expression of ZO-1 and ZO-2 on podocyte structure and function. Genes Cells, 23, 546-556.
- Jänne, P.A., Wozniak, A.J., Belani, C.P., Keohan, M.L., Ross, H.J., Polikoff, J.A., Mintzer, D.M., Ye, Z., Monberg, M.J. and Obasaju, C.K.; Pemetrexed expanded access program investigators. (2006): Pemetrexed alone or in combination with cisplatin in previously treated malignant pleural mesothelioma: outcomes from a phase IIIB expanded access program. J. Thorac. Oncol., 1, 506-512.
- Justice, R.W., Zilian, O., Woods, D.F., Noll, M. and Bryant, P.J. (1995): The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev., 9, 534-546.
- Kakiuchi-Kiyota, S., Schutten, M.M., Zhong, Y., Crawford, J.J. and Dey, A. (2019): Safety Considerations in the Development of Hippo Pathway Inhibitors in Cancers. Front. Cell Dev. Biol., 7, 156.
- Kaneda, A., Seike, T., Danjo, T., Nakajima, T., Otsubo, N., Yamaguchi, D., Tsuji, Y., Hamaguchi, K., Yasunaga, M., Nishiya, Y., Suzuki, M., Saito, J.I., Yatsunami, R., Nakamura, S., Sekido, Y. and Mori, K. (2020): The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res., 10, 4399-4415.
- Kim, Y.H., Goyal, M., Kurnit, D., Wharram, B., Wiggins, J., Holzman, L., Kershaw, D. and Wiggins, R. (2001): Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int., 60, 957-968.
- Lu, L., Li, Y., Kim, S.M., Bossuyt, W., Liu, P., Qiu, Q., Wang, Y., Halder, G., Finegold, M.J., Lee, J.S. and Johnson, R.L. (2010): Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA, 107, 1437-1442.
- Lu, L., Finegold, M.J. and Johnson, R.L. (2018): Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp. Mol. Med., 50, e423.
- Makita, R., Uchijima, Y., Nishiyama, K., Amano, T., Chen, Q., Takeuchi, T., Mitani, A., Nagase, T., Yatomi, Y., Aburatani, H., Nakagawa, O., Small, E.V., Cobo-Stark, P., Igarashi, P., Murakami, M., Tominaga, J., Sato, T., Asano, T., Kurihara, Y. and Kurihara, H. (2008): Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. -. Ren. Physiol., 294, 542-553.
- Mårtensson, J. and Bellomo, R. (2014): The rise and fall of NGAL in acute kidney injury. Blood Purif., 37, 304-310.
- Matsui, Y. and Lai, Z.C. (2013): Mutual regulation between Hippo signaling and actin cytoskeleton. Protein Cell, 4, 904-910.
- McClatchey, A.I., Saotome, I., Mercer, K., Crowley, D., Gusella, J.F., Bronson, R.T. and Jacks, T. (1998): Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev., 12, 1121-1133.
- Messmer, K.J. and Abel, S.R. (2001): Verteporfin for age-related macular degeneration. Ann. Pharmacother., 35, 1593-1598.
- Moroishi, T., Hansen, C.G. and Guan, K.L. (2015): The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer, 15, 73-79.
- Mundel, P. and Reiser, J. (2010): Proteinuria: an enzymatic disease of the podocyte? Kidney Int., 77, 571-580.
- Mundel, P., Heid, H.W., Mundel, T.M., Krüger, M., Reiser, J. and Kriz, W. (1997): Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol., 139, 193-204.
- Nan-ya, K., Kajihara, M., Kojima, N. and Degawa, M. (2015): Usefulness of urinary kidney injury molecule-1 (Kim-1) as a biomarker for cisplatin-induced sub-chronic kidney injury. J. Appl. Toxicol., 35, 124-132.
- Nardone, G., Oliver-De La Cruz, J., Vrbsky, J., Martini, C., Pribyl, J., Skládal, P., Pešl, M., Caluori, G., Pagliari, S., Martino, F., Maceckova, Z., Hajduch, M., Sanz-Garcia, A., Pugno, N.M., Stokin, G.B. and Forte, G. (2017): YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun., 8, 15321.
- Noland, C.L., Gierke, S., Schnier, P.D., Murray, J., Sandoval, W.N., Sagolla, M., Dey, A., Hannoush, R.N., Fairbrother, W.J. and Cunningham, C.N. (2016): Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure, 24, 179-186.
- Ozer, J.S., Dieterle, F., Troth, S., Perentes, E., Cordier, A., Verdes, P., Staedtler, F., Mahl, A., Grenet, O., Roth, D.R., Wahl, D., Legay, F., Holder, D., Erdos, Z., Vlasakova, K., Jin, H., Yu, Y., Muniappa, N., Forest, T., Clouse, H.K., Reynolds, S., Bailey, W.J., Thudium, D.T., Topper, M.J., Skopek, T.R., Sina, J.F., Glaab, W.E., Vonderscher, J., Maurer, G., Chibout, S.D., Sistare, F.D. and Gerhold, D.L. (2010): A panel of urinary biomarkers to monitor reversibility of renal injury and a serum marker with improved potential to assess renal function. Nat. Biotechnol., 28, 486-494.
- Park, J. and Hansen, C.G. (2021): Cellular feedback dynamics and multilevel regulation driven by the hippo pathway. Biochem. Soc. Trans., 49, 1515-1527.
- Pavenstädt, H., Kriz, W. and Kretzler, M. (2003): Cell biology of the glomerular podocyte. Physiol. Rev., 83, 253-307.
- Pei, T., Li, Y., Wang, J., Wang, H., Liang, Y., Shi, H., Sun, B., Yin, D., Sun, J., Song, R., Pan, S., Sun, Y., Jiang, H., Zheng, T. and Liu, L. (2015): YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget, 6, 17206-17220.
- Pfaller, W. and Gstraunthaler, G. (1998): Nephrotoxicity testing in vitro--what we know and what we need to know. Environ. Health Perspect., 106 (Suppl 2), 559-569.
- Piccolo, S., Dupont, S. and Cordenonsi, M. (2014): The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev., 94, 1287-1312.
- Prozialeck, W.C., VanDreel, A., Ackerman, C.D., Stock, I., Papaeliou, A., Yasmine, C., Wilson, K., Lamar, P.C., Sears, V.L., Gasiorowski, J.Z., DiNovo, K.M., Vaidya, V.S. and Edwards, J.R. (2016): Evaluation of cystatin C as an early biomarker of cadmium nephrotoxicity in the rat. Biometals, 29, 131-146.
- Rausch, V. and Hansen, C.G. (2020): The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol., 30, 32-48.
- Reginensi, A., Scott, R.P., Gregorieff, A., Bagherie-Lachidan, M., Chung, C., Lim, D.S., Pawson, T., Wrana, J. and McNeill, H. (2013): Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet., 9, e1003380.
- Reiser, J. and Altintas, M.M. (2016): Podocytes. F1000 Res., 5, 1-19.
- Ricardo, S.D., Bertram, J.F. and Ryan, G.B. (1994): Antioxidants protect podocyte foot processes in puromycin aminonucleoside-treated rats. J. Am. Soc. Nephrol., 4, 1974-1986.
- Schlegelmilch, K., Mohseni, M., Kirak, O., Pruszak, J., Rodriguez, J.R., Zhou, D., Kreger, B.T., Vasioukhin, V., Avruch, J., Brummelkamp, T.R. and Camargo, F.D. (2011): Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell, 144, 782-795.
- Schwartzman, M., Reginensi, A., Wong, J.S., Basgen, J.M., Meliambro, K., Nicholas, S.B., D’Agati, V., McNeill, H. and Campbell, K.N. (2016): Podocyte-Specific Deletion of Yes-Associated Protein Causes FSGS and Progressive Renal Failure. J. Am. Soc. Nephrol., 27, 216-226.
- Scott, R.P. and Quaggin, S.E. (2015): Review series: the cell biology of renal filtration. J. Cell Biol., 209, 199-210.
- Song, H., Mak, K.K., Topol, L., Yun, K., Hu, J., Garrett, L., Chen, Y., Park, O., Chang, J., Simpson, R.M., Wang, C.Y., Gao, B., Jiang, J. and Yang, Y. (2010): Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA, 107, 1431-1436.
- Srivastava, T., Garola, R.E., Whiting, J.M. and Alon, U.S. (2001): Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int., 59, 118-125.
- St John, M.A., Tao, W., Fei, X., Fukumoto, R., Carcangiu, M.L., Brownstein, D.G., Parlow, A.F., McGrath, J. and Xu, T. (1999): Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet., 21, 182-186.
- Thakur, V., Walker, P.D. and Shah, S.V. (1988): Evidence suggesting a role for hydroxyl radical in puromycin aminonucleoside-induced proteinuria. Kidney Int., 34, 494-499.
- Thielemans, N., Lauwerys, R. and Bernard, A. (1994): Competition between albumin and low-molecular-weight proteins for renal tubular uptake in experimental nephropathies. Nephron J., 66, 453-458.
- Torregrosa, I., Montoliu, C., Urios, A., Andrés-Costa, M.J., Giménez-Garzó, C., Juan, I., Puchades, M.J., Blasco, M.L., Carratalá, A., Sanjuán, R. and Miguel, A. (2015): Urinary KIM-1, NGAL and L-FABP for the diagnosis of AKI in patients with acute coronary syndrome or heart failure undergoing coronary angiography. Heart Vessels, 30, 703-711.
- Vaidya, V.S., Ozer, J.S., Dieterle, F., Collings, F.B., Ramirez, V., Troth, S., Muniappa, N., Thudium, D., Gerhold, D., Holder, D.J., Bobadilla, N.A., Marrer, E., Perentes, E., Cordier, A., Vonderscher, J., Maurer, G., Goering, P.L., Sistare, F.D. and Bonventre, J.V. (2010): Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol., 28, 478-485.
- Wang, S., Ma, K., Chen, L., Zhu, H., Liang, S., Liu, M. and Xu, N. (2016): TAZ promotes cell growth and inhibits Celastrol-induced cell apoptosis. Biosci. Rep., 36, 1-10.
- Wang, Y., Wang, Y.P., Tay, Y.C. and Harris, D.C. (2000): Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int., 58, 1797-1804.
- Wharram, B.L., Goyal, M., Wiggins, J.E., Sanden, S.K., Hussain, S., Filipiak, W.E., Saunders, T.L., Dysko, R.C., Kohno, K., Holzman, L.B. and Wiggins, R.C. (2005): Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol., 16, 2941-2952.
- Wong, J.S., Meliambro, K., Ray, J. and Campbell, K.N. (2016): Hippo signaling in the kidney: the good and the bad. Am. J. Physiol. Renal Physiol., 311, F241-F248.
- Wu, M.T., Lam, K.K., Lee, W.C., Hsu, K.T., Wu, C.H., Cheng, B.C., Ng, H.Y., Chi, P.J., Lee, Y.T. and Lee, C.T. (2012): Albuminuria, proteinuria, and urinary albumin to protein ratio in chronic kidney disease. J. Clin. Lab. Anal., 26, 82-92.
- Xin, M., Kim, Y., Sutherland, L.B., Murakami, M., Qi, X., McAnally, J., Porrello, E.R., Mahmoud, A.I., Tan, W., Shelton, J.M., Richardson, J.A., Sadek, H.A., Bassel-Duby, R. and Olson, E.N. (2013): Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA, 110, 13839-13844.
- Zhang, X., George, J., Deb, S., Degoutin, J.L., Takano, E.A., Fox, S.B., Bowtell, D.D. and Harvey, K.F.; AOCS Study group. (2011): The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene, 30, 2810-2822.
- Zhou, D., Conrad, C., Xia, F., Park, J.S., Payer, B., Yin, Y., Lauwers, G.Y., Thasler, W., Lee, J.T., Avruch, J. and Bardeesy, N. (2009): Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell, 16, 425-438.
- Zhou, Y., Huang, T., Cheng, A.S., Yu, J., Kang, W. and To, K.F. (2016): The TEAD family and its oncogenic role in promoting tumorigenesis. Int. J. Mol. Sci., 17, 1-15.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_175-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_175-t2.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_175-t3.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_175-t4.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)